Unshackling Development’s Handcuffs on Social Cognition
By Yu-Tzu Shih, Jason Alipio and Amar Sahay
Neurodevelopmental disorders arise in part from genetic mutations that disrupt formation of brain circuits. Loss of one copy of the Dyrk1a gene, which encodes an enzyme called dual specificity tyrosine-phosphorylation-regulated kinase 1A, results in a syndromic form of autism –a neurodevelopmental disorder that is characterized by impaired social cognition, repetitive behavior and increased seizure risk (https://gene.sfari.org/database/human-gene/DYRK1A). Dyrk1a is ubiquitously expressed in the brain and phosphorylates more than 30 different substrates, many of which are selectively expressed in specific cell-types. Through recruitment of different substrates, DYRK1A regulates diverse biological processes such as cell cycle, neuronal migration, axonal and dendritic development, and synapse formation. The synaptic and circuit mechanisms by which DYRK1A contributes to social cognition are poorly understood.
Currently, there are no treatments to reverse Dyrk1a-associated circuit and cognitive impairments.
Two major challenges to overcome when devising strategies to rescue phenotypes associated with Dyrk1a heterozygosity are determining when to intervene and exerting cell-type specificity. With regards to timing, the window of therapeutic intervention ideally is after birth, either during the postnatal period when brain circuits undergo refinement or in adulthood, if experience-sensitive plasticity mechanisms can still be harnessed to restore circuit functions. The second challenge is to avoid activation of DYRK1A in all cell-types since this poses a cancer risk and harbors potential to disrupt many cellular functions that are dependent on DYRK1A substrates.
Growing evidence suggests a role for parvalbumin inhibitory neurons (PV INs) in pathology of autism spectrum disorder. In prior work, we had identified ABLIM3, a cytoskeletal protein that functions as a brake on experience-dependent regulation of PV IN mediated GABAergic inhibition in the hippocampus, a brain region critical for forming memories. Here, we demonstrated that ABLIM3 is a synaptic substrate of DYRK1A in hippocampal mossy fiber-PV IN synapses. We found that in wild type mice, social experience causes a decrease in ABLIM3 in hippocampal mossy fibers, resulting in increased excitatory drive onto parvalbumin inhibitory neurons and increasing GABAergic inhibition of downstream principal cells. Further, we showed that in adult Dyrk1a heterozygous mice, ABLIM3 levels fail to decrease in response to social experience and this prevents experience-dependent recruitment of PV IN mediated GABAergic inhibition of principal cells.
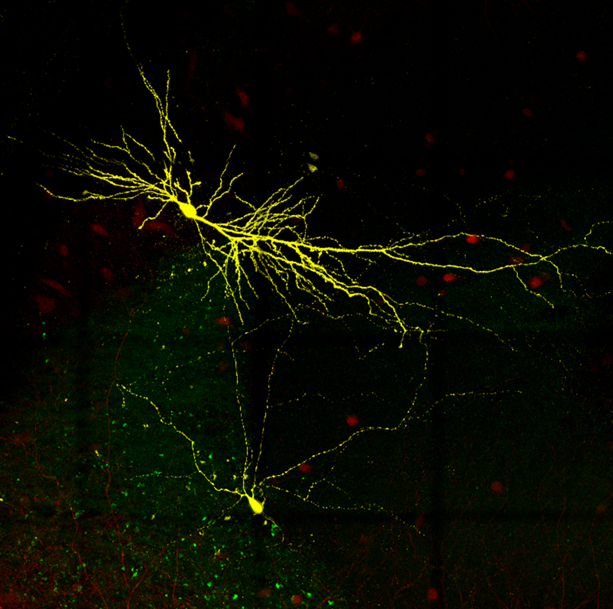
Parvalbumin interneuron and pyramidal cell in hippocampal CA2.
Using viral gene therapy, we showed that downregulating ABLIM3 in hippocampus of adult Dyrk1a heterozygous mice was sufficient to restore GABAergic inhibition and social cognition. Direct activation of parvalbumin interneurons in the hippocampus of adult Dyrk1a heterozygous mice also rescued social cognition. Together these findings demonstrate that targeting a synaptic substrate of DYRK1A in the hippocampus of adult mice is an effective way to rescue Dyrk1A-associated developmental impairments in GABAergic inhibition and social cognition.
Ongoing research in our lab supports the thesis that a significant number of ultra-high confidence psychiatric disease risk genes converge upon regulation of experience-dependent parvalbumin interneuron plasticity in the hippocampus. However, experience-dependent parvalbumin interneuron plasticity is central to the dynamic regulation of perisomatic inhibition of principal cells in not just hippocampus but also in other brain regions supporting cognitive flexibility, decision making and attention. Ultimately, we hope that our studies will guide therapeutic strategies to restore experience-dependent parvalbumin interneuron plasticity, impact different domains of cognition and reduce seizure risk in neurodevelopmental disorders. Moving the needle towards this goal will have a profound influence on patients’ lives.
Yu-Tzu Shih and Jason Alipio are postdoctoral trainees in the Sahay laboratory.
Amar Sahay is a Professor of Psychiatry at Harvard Medical School and Principal Faculty member of the Harvard Stem Cell Institute, and Massachusetts General Hospital, as well as an Associate Member of the Broad Institute.
Learn more in the original research article:
An inhibitory circuit-based enhancer of DYRK1A function reverses Dyrk1a-associated impairment in social recognition.
Shih YT, Alipio JB, Sahay A. Neuron. 2023 Oct 4;111(19):3084-3101.e5.
News Types: Community Stories